The minimal goal of any RNA-seq experiment is to generate actionable data. Commonly accepted RNA-seq analysis pipelines usually include an adapter clipping step which removes (or masks) read bases that match adapter sequence. In the last 6 months, with increased use of our NextSeq (at 2×150 bp), we have observed interesting clipping anomalies with some of our RNA-seq libraries, where a large number of bases are clipped from the final data output. One might ask, “why would adapter sequence show up in the data?” And, this is a fair question. Interestingly, the answer is not what you might expect. Thus, we wanted to bring our findings to light here.
The vast majority of commercially produced RNA-Seq kits are designed to generate NGS libraries with an insert size distribution centered around 150 bp or so. This size is not intuitively obvious from final library QC (i.e. Bioanalyzer or Tapestation analysis). Shown below is a typical bioanalyzer trace of a stranded mRNA Seq library using a commercially available kit.
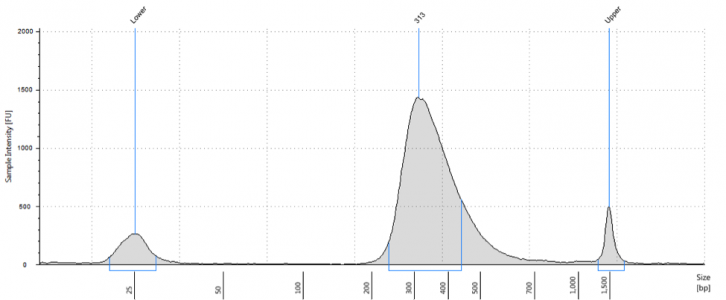
The apparent average library size is 313 bp. It’s important to remember that the average is 313 bp but there is a distribution of sizes ranging from 200-500 bp. Importantly, the Illumina adapters add around 125 bp total sequence to the final library length. Thus, average insert size is 300-125= 175 bp, with a range of sizes from 75-375bp. Keep in mind that there is a slight bias for smaller fragments during cluster generation, so these fragments will not have an even distribution on the flow cell. Regardless, these libraries will sequence just fine using the 50 or 100 bp read lengths on the HiSeq, but what happens when your experimental question requires a little more coverage of the fragments (e.g. isoform detection or transcriptome assembly)? Maybe you decide to put them on the MiSeq for a 150, 250, or even 300 bp read length. Perhaps you need a fast turnaround time and opt for the NextSeq’s PE150 read capability.
If you were to perform a PE150 sequencing run with these libraries, you would start sequencing into adapter for any library molecules smaller than 150 bp. This is wasted sequencing!
Now, let’s consider, from a math perspective, how the fragments produced by a current mRNA-seq protocol are interacting with 2×150 base (paired-end 150 bp reads) read lengths. If you consider an average insert size of 175 bp, depending on the distribution (and for example sake let’s say is Poisson-like), approximately 25% of the fragments would be 150 bases or smaller. We actually see clipping in about half of the reads generated, which means the distribution is wider or the average is slightly lower than reported on the QC device.
Ok, let’s move from theoretical to empirical. Below you will see a bioanalyzer trace of cDNA after fragmentation. The fragmentation was performed per the protocol from the RNA-seq kit manufacturer. As you can see, there are a large number of fragments below 150 bp. When sequencing this library using longer paired-end reads (ex. 2×150), these fragments smaller than 150 bp will have adapter bases show up in the forward and reverse reads. This data will be clipped (red x’ed out areas). Thus, one will be losing actionable data from their RNA-seq experiment.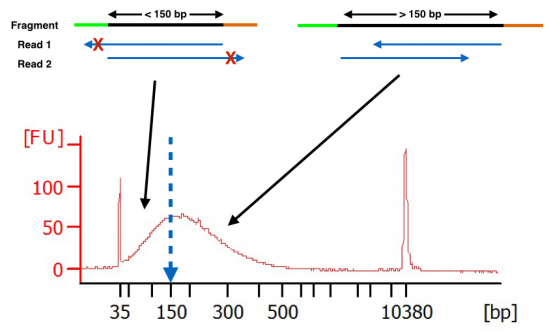
As a side note, this design does not work very well either for isoform detection with an isoform analysis pipeline, because you are essentially covering the same transcript fragment with both reads. The best case scenario for isoform detection would be to cover as many exon boundaries as possible in each transcript by using even longer reads with something like the MiSeq platform. Good luck using the protocol above as read lengths greater than about 100 bases will provide diminishing returns.
This is why we talk about RNA-seq for comparative expression and isoform detection as fundamentally two different experiments. This is not to say that isoform information cannot be gained from 2×100 or 2×150 bp data, however if that is your primary objective for RNA-seq, it might be best to approach it from a different molecular and platform angle. If you would like to learn more about the different approaches for differential expression and isoform detection, please see our earlier blogs on single and paired-end read data with RNA-seq.
Ok, so what can be done? Well, one solution would be to limit your read length for RNA-seq to a maximum of 100 bases…. This is unacceptable because hardware manufacturers will continue to develop longer and more accurate reads. The kit manufactureres (you know who you are) could have their groups hit the bench and publish some updated fragmentation conditions as a protocol addendum…. This is totally acceptable and the best case scenario, since it would then be supported by the companies and their technical support. We won’t hold our breaths for this one. So, the best solution for Cofactor (and for our clients) was to go back to the drawing board and fine tune fragmentation conditions and develop our own protocol addendum. Whether you are fragmenting RNA by chemical means, or cDNA by mechanical or enzymatic means, it is imperative that these steps be optimized to suit your longest experimental design/desired read length. This turns out to be no easy task, as things like sample quality and quantity can have dramatic effects on your ability to generate ideal fragment sizes.
Luckily, we actually enjoy spending hours in the lab trouble-shooting and optimizing conditions for different RNA-Seq workflows and read lengths. It is extremely satisfying to us lab folks when we are able to help our clients get the most out of their NGS projects. If you are considering an RNA Seq project, contact one of our project scientists to hear how we can customize insert size to best fit your experimental goals. We want to make sure you are getting the most out of your sequencing data.