Part 2 of our series on RNA-seq. Part 1, Part 3
2. Poly(A) enrichment or ribosomal removal?
To continue where we left off yesterday, I sometimes see researchers fretting over whether to do poly(A) enrichment or ribosomal removal for their samples prior to library construction. It is important to remember that each one will have some effect on the cost of the experiment and the data generated. Obviously (or not so obviously) this question usually arises with eukaryotes, which have polyadenylated (poly(A)) transcripts. In general, we find at Cofactor that we need to generate additional sequencing reads for eukaryotic samples where we perform ribosomal reduction as opposed to poly(A) enrichment. There will be additional transcripts that come along for the ride following ribosomal removal (lncRNAs, some fraction of incompletely-transcribed mRNAs will be partially-spliced or un-spliced but not polyadenylated, and spliced but undegraded introns may show up as well that don’t show up in poly(A) enriched samples [1]). Since additional transcripts, and material, make it through the process, we have to try and account for these when determining the number of reads needed for an experiment. Often it is advantageous to over sequence several samples first, generate saturation curves for these samples (shown below), and determine the best level of sequencing, while accounting for the percentage of ribosomal reads, for follow-up samples.
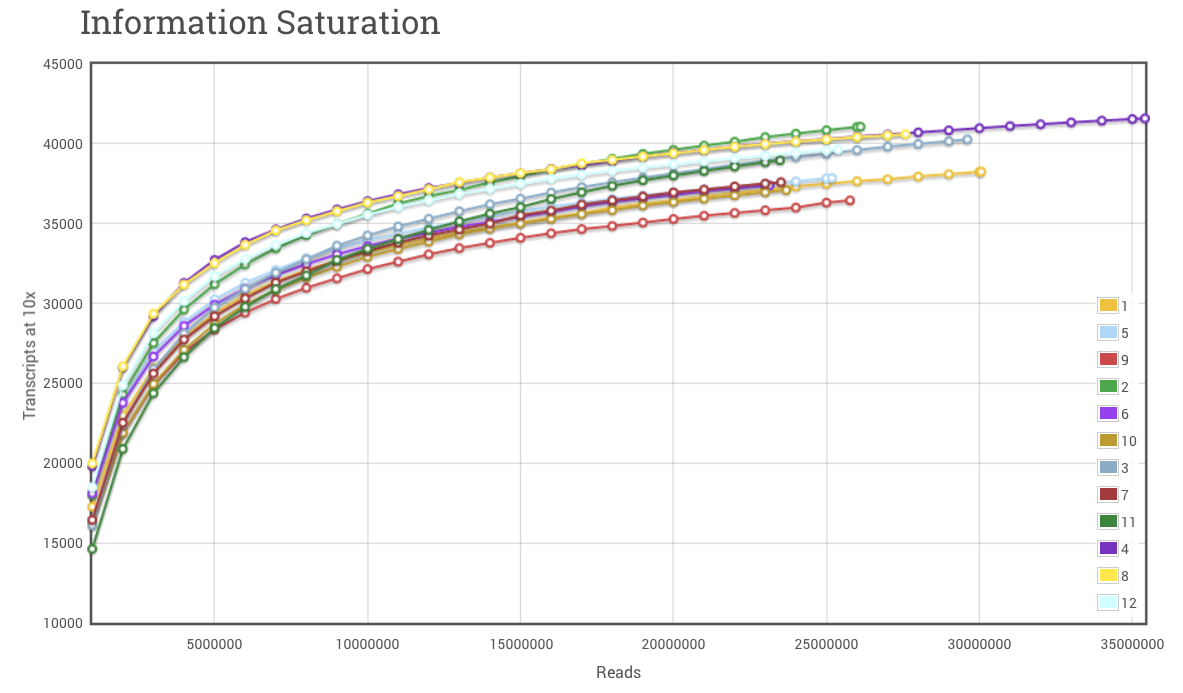
Figure 1. RNA-seq Saturation Curve. The saturation curve above displays the number of transcript references with < 10 read alignments at bins of 1 million reads. As additional reads are cumulatively added to the alignment set, hits to transcript references increase very rapidly at first, and then level off to become asymptotic to the upper limit. The upper limit in RNA-seq experiments will be defined by several factors, not limited to, number of maximum sequencing reads, number of transcript/isoform references and complexity of the original RNA pool.
With poly(A) samples, sequencing estimations are much more straightforward, as Cofactor has historical saturation data on human, mouse, rat, CHO, algae, fungal, bacterial, and many, many more organisms to help answer the question, “how many reads do I need for my experiment?”
Interestingly, there are organisms that are not so straightforward, such as algae, where ribosomal removal or poly(A) enrichment could be used. And, also begs the question, “which is the best ribosomal kit to use? Plant? Animal? Bacterial?” In this case, where the question’s not easily answered and a google search doesn’t give up the glory in the first page or two, it may be best to just use poly(A) enrichment and accept 10-15% ribosomal reads.
One last point to note, there are several new interesting products that may be employed:
1) Epicentre has a new online tool out to help figure out which of their kits is the best match for ribo-depletion of non-standard organisms.
2) The other option is this new technology from NuGen that lets you deplete specific transcripts in RNA-seq. Here’s the blurb from their webpage.
InDA-C (Insert Dependent Adapter Cleavage) — the novel and NuGEN-proprietary InDA-C method employs customized probes during library creation to target specific transcript species for exclusion in RNA-Seq libraries. Unlike methods that use hybridization-mediated pull-down strategies to deplete unwanted RNA species prior to cDNA synthesis, the InDA-C method selectively targets and eliminates unwanted transcripts from finished libraries, avoiding potential off-target mRNA cross-hybridization events that have been demonstrated to introduce unwanted bias. The InDA-C method is currently employed in the Encore Complete Prokaryotic Library Systems to reduce ribosomal RNA transcripts and in the Ovation Human Blood RNA-Seq Library Systems to reduce both human globin and ribosomal RNA transcripts.
1. Morlan JD, Qu K, Sinicropi DV (2012) Selective Depletion of rRNA Enables Whole Transcriptome Profiling of Archival Fixed Tissue. PLoS ONE 7(8): e42882.doi: 10.1371/journal.pone.0042882