
What defines success for your RNA-seq experiment? It’s a question that we ask a lot around here (aside from “what are you doing for lunch”?). And what are the metrics we can use to answer that question? Making a bunch of A’s, T’s, G’s and C’s is only part of it. If you are new to our blog you may want to get up to speed and check out some of our other blog posts that will be relevant: saturation curves, spike-ins, and replicates (You need them if you want p-values). Today we will cover an often overlooked aspect of an RNA-Seq experiment: the choice of reference.
First, what is a reference? Simply put, a reference is a representation of the genes or transcripts in an organism. RNA-Seq measures the expression levels of transcripts, and to determine those levels all the reads must be aligned to an already assembled and annotated set of reference sequences. The choice of reference defines the lens through which the RNA-Seq data is viewed.
While it may seem straightforward to identify the coding sequences in a genome, gene sets can differ considerably, even in the human genome — not just in exon boundary placement but in how many transcripts there are. For example, UCSC’s version of the human gene set defines about 80,000 transcripts, whereas Ensembl’s has over 170,000.
Zhao and Zhang showed recently that aligning to different reference gene sets can influence the outcome of RNA-seq experiments. Looking at the genes shared among Ensembl, UCSC, and RefGene versions, they found that “…identical counts were obtained for only 16.3% of genes”.
To illustrate this further, we took one of our own samples, aligned it to both the UCSC and Ensembl gene sets, and plotted the results 1.
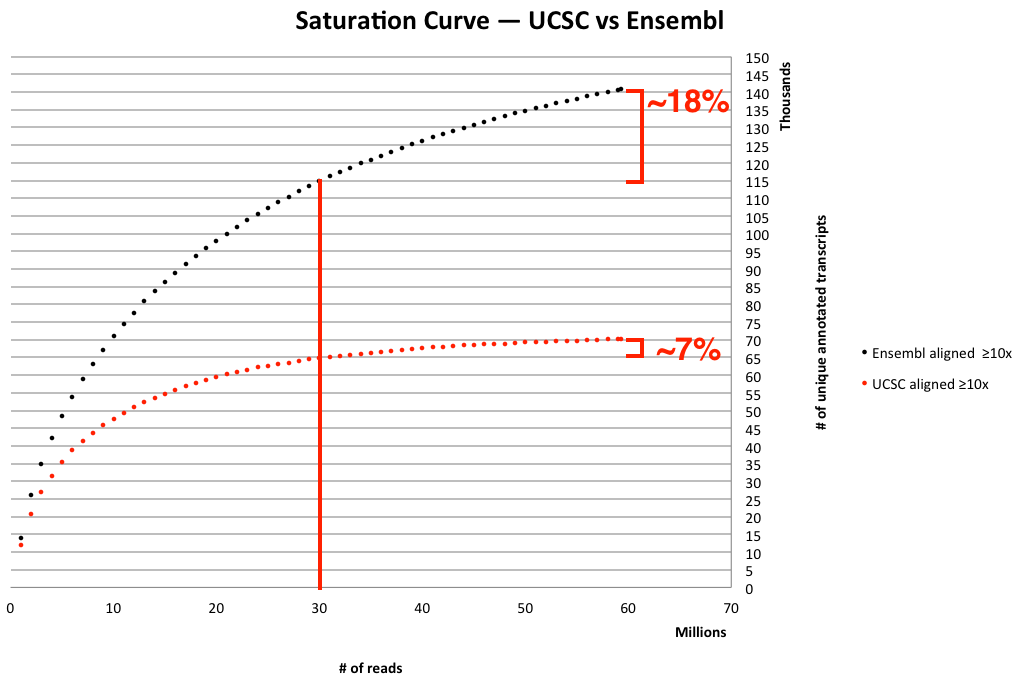
We took a single sample that was sequenced to around 60 million single-end reads and aligned to the UCSC transcript set and the Ensembl transcript set. As you can see, there is a large difference in the number of unique transcripts identified 2. Ensembl annotates more transcripts than UCSC and therefore requires a lot more reads to sample them at a “sufficient” amount.
Lets take a closer look at the graph. If we were to sequence 30 million reads (see the red vertical line) we may not sufficiently sample all the transcripts in the Ensembl database, but it looks like we sampled almost ~93% of all the transcripts annotated in the UCSC reference — the UCSC curve is already pretty flat at 30 million reads. By sequencing ~60 million reads, we’ve doubled the amount of data (and significantly increased the budget), but all that additional data bring only a ~7 % increase in new information gained. The extra data helps even more when aligning to Ensembl, bringing an 18% increase in unique transcripts seen.
So what does this mean for your research? If your goal is discovery — casting the widest net — then using the larger set of transcripts as annotated in the Ensembl reference set may make more sense. Just keep in mind the increase in data (and money) required to sample those transcripts effectively (see graph above). This may impact your budget dramatically enough to make multiple samples and replicates not fiscally possible. Conversely, you may choose a more conservative 3 reference like the UCSC transcripts set and sequence multiple replicates at 30 million reads. For the same money, you’d gain significant statistical power.
As you can see, there are some important considerations before you even pick up a Pipetman. Here are some questions I ask the scientists we work with:
- What reference sequence have you used in the past? This may make the decision for you if you want to have your new dataset comparable to older ones. Or you may want to take a look at your data against a different reference that has a richer set of annotated isoforms or transcripts not identified in your previous study.
- If you are looking for a transcript(s) a priori in your data set, be sure it’s in the gene set you are aligning to.
- Do I have enough read counts to reliably identify the transcripts I’m interested in, i.e. low abundance/multiple annotated iso-forms? How will this impact the sensitivity of my experiment?
Understanding the goals — what defines a successful experiment — is an important part of choosing a reference. If you are planning any RNA-seq experiments, reach out to one of our project scientists to discuss your project and goals.
- Thank you to our awesome computational scientist Ajay Khanna for pulling the data for me to make this graph. ↩
- Note that here we require a transcript to have at least 10 reads align to it. ↩
- By conservative I mean that the UCSC transcript set is more stringent in its requirements to include transcripts into its reference. ↩
