I have conversations quite often where an individual expresses interest in using less and less DNA for next-generation sequencing library construction. Of note, most of their interest surrounds disease samples where the material may be limiting and of a heterogenous nature. Interestingly, many times during the course of these conversations, the individual also states that they are looking to detect lower and lower minor allele frequencies (MAFs) as well as lowering the DNA input. I have not seen this topic covered in-depth before, so I wanted to discuss the balance between the amount (or copies) of DNA going into the library process, sequence coverage generated and the lower theoretical threshold of minor allele frequency detection.
When heterogenous DNA material is prepped, sequenced and variants are called from the resulting data, allele percentages do not follow normal patterns of germline heterozygous and homozygous variant percentages seen with DNA of higher homogeneity. Instead, we see smaller (in some cases, much smaller) percentages of alternate somatic alleles in the data, which are referred to as minor alleles. The minor allele contribution to the whole is termed minor allele frequency (MAF).
At Cofactor, a heterogenous sample represents any groups of cells or DNA that has multiple species of cell or DNA type (e.g. what one might see from a tumor biopsy sample). In most cases, the DNA we work with is cancer DNA extracted from fresh-frozen, FFPE or cell-free material and currently there exists three main ways to interrogate genomic variation in these samples, including whole-genome sequencing (WGS), whole-exome sequencing (WES), custom hybrid capture sequencing (using custom capture panels) and targeted PCR amplicon sequencing. At the current price per base, WGS is not routinely employed for MAF detection in heterogenous tissue (however let’s not rule out future applications on the Illumina X10). It would simply be too expensive. The most common and widely used methods are WES, custom capture panels, and targeted amplicon sequencing. For arguments sake, let’s take a look at the widely used application of WES and custom capture panels.
In our process, once library construction, captures and sequencing is complete, we move the data into our variant calling pipeline, which uses a deduplication module to remove groups of paired-end reads that share the same start sites with other pairs (take note, this is a super-important point of difference between capture versus PCR based enrichment) . Once removed, the resulting data more accurately represents the genomic areas of interest in the sample. We are able to confidently remove these PCR duplicates, produced during the molecular procedures, because any two groups of read pairs that share the same sequencing start sites most likely arose from PCR copies produced from the same adapter ligated capture fragment. The ultimate goal is to call and determine frequencies of alleles from uniquely sequenced genomic fragments while removing duplicate sequence data. Deduplication is awesome because it helps to mitigate potential skew in MAFs due to the inherent variability in the PCR amplification steps required during library prep.
Ok, so what does this all mean and why does it matter? Well, I plan on talking about MAF, so lets move into that now, however I will be speaking below in the context of capture-based experiments and not PCR-based experiments. Also, I won’t be commenting on samples where whole-genome amplification (WGA) was performed prior to capture. This adds an additional layer of complexity that I may cover in another blog post. Some of the math and concepts I present below are easier to pick up when they represent sequencing data derived from unique genomic fragments that we get out of routine capture experiments (non-WGA samples).
Quick note; there are numerous groups working with ultra-low imput DNA library kits to generate material for WES/custom capture panels and variant detection in cancer samples. I am going to describe how, below defined DNA input amounts, this strategy will not provide the desired MAF sensitivity.
On to the nitty-gritty!!
The basic essential component driving MAF sensitivity is the number of sequences that cover the base of interest, most simply termed sequence coverage. If we have a maximum of 100,000x coverage following deduplication, and require that at least 4 reads show the alternate allele to make a SNP call, the best MAF we can derive is 0.004%. Which, if we could reliably detect MAF’s down to this point, we would be considered rock stars!!! However, we have machine noise, molecular noise (polymerase error rate), and variability driven by the capture procedure that plays a major role in keeping us from ever reaching the lowest theoretical MAF. Therefore, let’s say a log higher will get us over noise and we use 40 reads for a 0.04% MAF. This is awesome too!!! But wait….let’s look at it from another angle. No one routinely sequences to 100,000x coverage. Coverage is not only the main driving factor for MAF, but also the main limitation. Let’s put this level of coverage into perspective:
Application Amount of Data for 100k coverage
Whole Genome sequencing (3 Gb) 300 Tb
Exome capture (50 mb) 5 Tb
The math used to derive the above numbers is simple; Coverage x Total Bases Sequenced = Bases of Data Required.
For the exome capture; 50 Mb total capture bases x 100,00x coverage = 5 Tb of sequencing data required (with paired-end 150 bp reads, that would be 333 BILLION Reads)
You probably already thought to yourself after seeing the above numbers, “this is just not gonna work”! No one is going to pay to sequence 5 Tb for exome studies (not yet at least, maybe in the next two years we can get that much data cost effectively).
A good follow-on question would be, “To what level of coverage do I need to target for the sensitivity I require”? Great question and let’s figure that out for something like a 100 kb custom capture panel.
We want to detect things pretty confidently to 0.1% sensitivity while requiring 4 reads to show the alternate allele.
4 reads / 0.001 = 4000x coverage.
We have performed these experiments at Cofactor using custom capture panels, and the numbers don’t track exactly, but good enough to warrant a very close look at the data. We found some very interesting anomalies at this level concerning false-positive variant calls and are able to now filter and correct for them (yet, still are not able to reach the theoretical maximum MAF sensitivity).
So, we covered one of the easier concepts, coverage is a driver of MAF sensitivity.
Next, let’s look at the MAF from a different perspective… copies of DNA, specifically capture experiments with limiting DNA material. As I have mentioned several times; capture experiments are excellent applications to interrogate MAF’s because we are able to remove duplicate reads and distill our data down to reads originating from unique genomic fragments. If we find that we need to generate 4000x coverage to drive a lower sensitivity of 0.1% MAF, and ultimately this coverage would be derived from deduplicated data, how many genome copies would we need? In a perfect world, we would need at least 4000 haploid genome copies, or 2000 cells, or 12 ng of DNA. In other words, no matter how many sequencing reads you generate, you can never attain a greater deduplicated coverage than the number of genomics copies going onto the sequencer (I am talking about samples that have not gone through whole-genome amplification). Now, hold on for a minute. At Cofactor we actually see these numbers increase by a log (10x), due to process noise, variability inherent with the solution-based capture and PCR amplifications and loss during the molecular procedures (plus, we like to be conservative here). Taking this into account, for 4000x unique coverage, we would need about 20,000 cells or 120 ng of DNA. Remember, these are maximum numbers and even with these amounts, we cannot quite get down to 0.1% for most variant calls due to the poisson distribution of coverage (check out poisson distribution of coverage here). The table below shows the maximum theoretical coverage and lowest MAF sensitivity for different numbers of cells, DNA input and haploid genomes. Remember, these are theoretical, perfect-world numbers and I show them for description only.
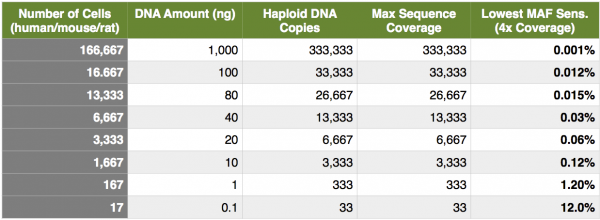
Easily surmised from above, but not easily surmised from some of my conversations, is that kits allow you to input lower and lower amounts of DNA at the expense of an INCREASING MAF sensitivity! Let’s say that Genome International (amazingly inventive name… thank you!) released a kit with a lower input of 1 ng of native (non-amped) DNA. You and your colleagues were really excited to use this kit, prior to a custom capture panel, on your fine needle aspirates to detect MAF down to 0.1%. If you look at the table above, you can see even under perfect circumstances, this is IMPOSSIBLE!. You will not be able to even generate the molecules from the native library prep to drive the coverage needed to call variants at a theoretical sensitivity of 0.1%, at a theoretical best 1.2% (and probably more like 12% taking all variability and noise into account). Second, taking coverage into consideration, you would HAVE TO sequence to saturation, meaning you sequenced every unique molecule from the library at least once. Take home point is, a balance exists between the number of DNA copies going into library construction and capture, the amount of sequence data generated, and the lowest confident MAF sensitivity.
To close, let’s consider a question I emailed to all of the folks at Cofactor several months back (with the offering that I would buy lunch for anyone with the correct answer).
When using 15 ng of cfDNA for library construction and capture, what is the highest theoretical maximum sequence coverage that could be attained at any position? What is the lowest theoretical minor allele frequency (MAF) we could detect?
First, we need to set several parameters:
Minimum 4 supporting reads to call a variant
Both strands need to be represented in the SNP calls.
6 pg DNA for diploid cell (mouse/human/rat)
Ok, now stop reading here, grab a pen, paper and calculator and work it out……
Finished… get your answer ready for orthogonal validation!
I show here the correct answer received from Cofactor’s winner, Dr. Dave Messina, our esteemed Director of bioinformatics (big surprise, that guy is a genius!).
15 ng total DNA / 6 pg per cell = 2500 cells worth of DNA per capture.
2500 cells * 2 copies per cell = 5000 haploid genomes in 15 ng captured = 5000 maximum theoretical depth.
4 reads minimum / 5000 max depth = 0.08% lowest theoretical minor allele frequency.
Taking all of the above into account, you would really need to sequence to somewhere around 10,000x unique coverage, sequence to saturation, and have a really killer variant call pipeline with stringent false-positive filtering.
In summary, much of the above discussion covers theoretical coverage maximums and lowest MAF sensitivities based on variant calls in non-amplified and capture enriched DNA samples. Once we take process noise, molecular variability and coverage distributions into account, you would probably need to increase the numbers in columns 1-4 in the table above by 10-fold while tweaking your molecular and variant call pipelines. If you want to have the best chance of reaching the lowest theoretical lowest MAF sensitivity, you should use the most DNA material possible for library construction, sequence to the highest coverage your budget can handle, and be ready to stringently filter your variant calls for false positives (after deduplication). As an added note, you should also investigate the level of sequence saturation attained.
Again, thanks for reading and please let us know if you have any questions. We appreciate questions and comments!
Until next time, copies = coverage = MAF sensitivity.