Part 3 of our series on RNA-seq. Part 1, Part 2
3. Use spike-in controls
Spike-in controls, whether custom, or the ERCC set can be an extremely useful tool, even they are only used for assessing the quality and success of your library construction process and reagents. At the beginning of a transcriptome or RNA-seq experiment, everything in the pool is an unknown and potentially variable (including what are believed to be molecular controls such as housekeeping genes). Spike-ins supply a number of transcripts at known concentrations, lengths and sequence, and since they are added at the very beginning serve as a great control. When used as a library QC, if at the end of sequencing all of the reads are from the spike-in controls, you should hold your sample quality/quantity highly suspect. On the other hand, if you finish the library construction process and there is not a positive signal following PCR, I would hold the library preparation procedure, and potentially the kit, suspect.
Spike-in controls are inherently advantageous to endogenous housekeeping genes for normalization, as potential housekeeping genes such as ACTB, GAPDH, HPRT1, and B2M, etc. vary considerably under different experimental conditions [1-3]. An additional advantage of spike in controls, over endogenous controls, when used for normalization is that there are between 48-96. This number is much greater than the number of unchanging endogenous genes that would be used for normalization. Thus, potential error associated with normalization based on few endogenous genes is minimized. At Cofactor, our belief is that the best and most effective way to normalize RNA-seq samples is by using spike in controls.
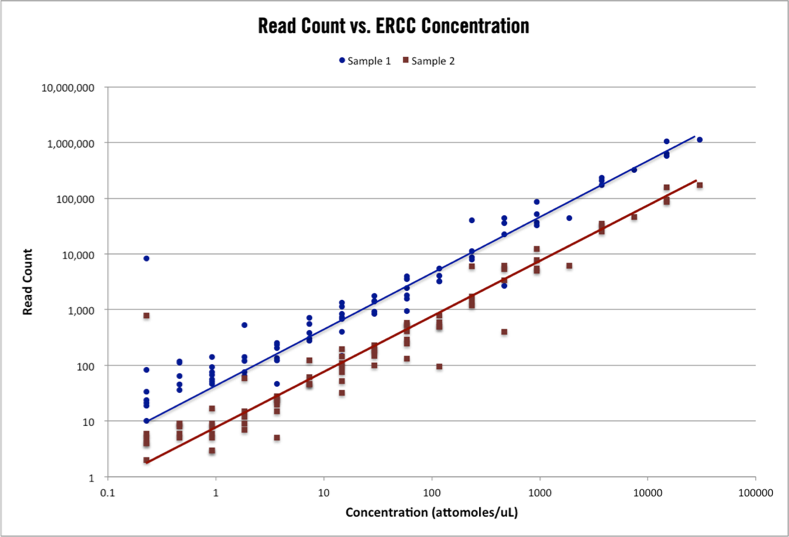 Figure 1 – ERCC control read counts. The above plot shows the read count alignments for 96 ERCC control transcripts in two RNA-seq samples with different total read counts. The range of ERCC control concentration is 10^5 and the topology of counts for each sample is nearly identical.
Figure 1 – ERCC control read counts. The above plot shows the read count alignments for 96 ERCC control transcripts in two RNA-seq samples with different total read counts. The range of ERCC control concentration is 10^5 and the topology of counts for each sample is nearly identical.
Cofactor works a bunch on low-input RNA, whether from FFPE material or other low content sources, and has been thinking a lot about the implementation of spike-ins with amplified RNA. As opposed to normal RNA sources, where we just get our concentrations, balance the total amount per sample and add the spike-ins, amplified RNA sources are much trickier. First, you have to have a pretty good handle on your concentration and total amount of RNA (Refer to Part 1 if you are wondering about the best way to deduce concentration). However, it is extremely hard to land on a confident number with many of the low-input samples we work with. Therefore, we will add spike-ins to serve as library controls, which is extremely helpful anyway with low input samples that will undergo amplification, but will not use the spike-ins for normalization procedures. As a side note, if you are wondering what’s our amplification kit “du-jour”, we have recently used the Clonetech SMART and NuGen Ovation kits, with a preference for the NuGen for it’s ease of use.
Until tomorrow….when we segway into analysis considerations.
We’re just getting started!
Jon
1. Rubie C, Kempf K, Hans J, Su T, Tilton B et al. (2005) Housekeeping gene variability in normal and cancerous colorectal, pancreatic, esophageal, gastric and hepatic tissues. Mol Cell Probes 19: 101-109.
2. Silver N, Best S, Jiang J, Thein SL. (2006) Selection of housekeeping genes for gene expression studies in human reticulocytes using real-time PCR. BMC Mol Biol 7:33.
3. de Jonge HJ, Fehrmann RS, Bont ES, Hofstra RM, Gerbens F, Kamps WA, de Vries EG, van der Zee AG, te Meerman GJ, ter Elst A. (2007) Evidence based selection of housekeeping genes. PLoS One 15(9):e898.