Those that know me well know that I am all about the interface between the genomics and the phenotype, the dynamic part of the biological system referred to in our industry as functional genomics. How this system behaves as a result of the underlying genomics and how it changes in response to environment, infection, and even treatment approaches is what gets me jazzed. Not surprisingly, Cofactor Genomics has created an entire business focused on standardizing functional genomics (namely RNA-seq) in order to be able to make some solid conclusions from studies using our products and services. This focus started in earnest 3 years ago when our large Pharma clients said they did not trust RNA-seq because there were no standards and essentially no way to judge a successful RNA-seq experiment from a failed. We spent the next three years in the lab and with our software developers to change that.
Given my interests, it is probably no surprise that most large scale population genomics studies have been what I would pick up to battle insomnia. I remember in graduate school that population genomics was on the other end of the spectrum of what I was interested in. I preferred learning about specific base changes in given genes that changed a pathway and resulted in an interesting phenotype. Despite my tendency away from them, this month a population genomics study was released that I could not put down.
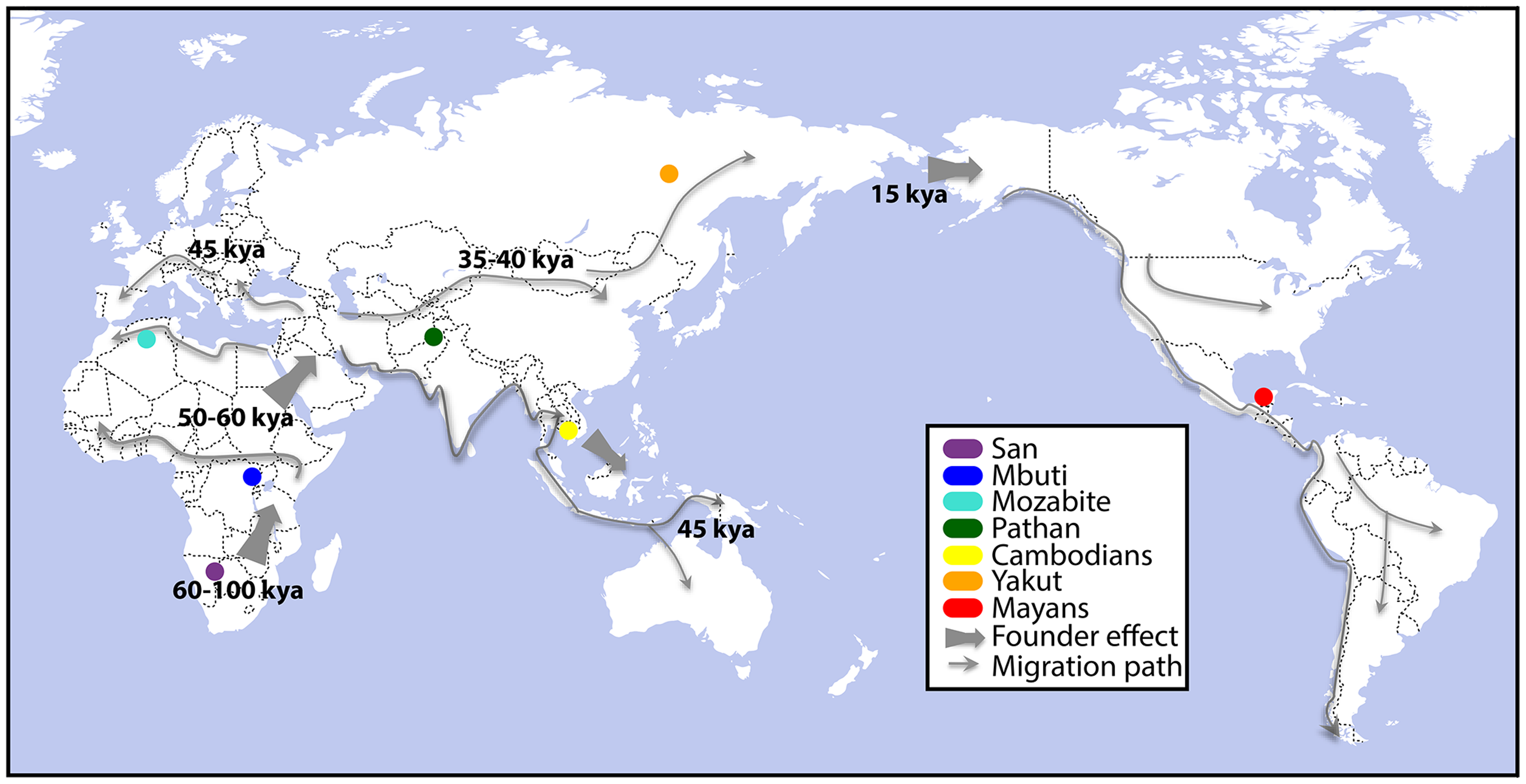
This month in PLoS Genetics a collaboration of many talented scientists (lead author Alicia Martin and last Author being Carlos Bustamante) took a functional genomics approach to population genetics in a publication focused on how differential expression of genes and isoforms are observed in different populations. The 7 populations that were the focus of the study are shown in the figure. Although the majority of the variation in expression that they witnessed was explained as normal variation within and between populations, there was an interesting set of about a dozen genes whose differential expression observation was population specific. Among genes showing differential gene expression were ones involved in immune response to influenza, West Nile virus, the avian flu, and other DNA and RNA viruses (MX1). There were also genes that have been associated with breast cancer risk in Europeans (LSP1). One final gene that caught my attention, was one implicated in acute myeloid leukemia (AML). AML was the subject of the last publication for which I was involved in at The Genome Institute before founding Cofactor Genomics in 2008, and represented a turning point for the Center/Institute when it changed its focus from genomic sequencing and assembly of genomes to applied genomics in the field of cancer. I found this list of genes particularly interesting because of the implications it had to different populations (and individuals) exhibiting characteristics of disease and/or infection susceptibility through the characterization of their RNA expression levels. This characterization of the functional genomics gets us one step closer to the phenotypes I find so fascinating.
There are a number of interesting findings from this pub I have not touched on (namely allele specific expression, promoters, and many of the methods the authors employed to achieve the results). I am just now diving into the supplementary material, but a congratulations to the authors on this functional genomics population study.
References: Martin A., Costa H., et. al. Transcriptome Sequencing from Diverse Human Populations Reveals Differentiated Regulatory Architecture. PLoS Genetics. 2014 August 14th; 10.1371/journal.pgen.1004549
Ley TJ, et. al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome.
Nature. 2008 Nov 6; 456(7218):66-72.