This month much of the discussion in the RNA-Seq world has centered on a group of papers just published in Nature Biotechnology. Two large research consortia and affiliated research teams undertook large-scale RNA-Seq studies evaluating the major sequencing platforms, library protocols, normalization methods, and spike-in controls. Taken together, the papers establish the current state of the field, determining the true reliability and reproducibility of RNA-Seq results using real-world data.
What are the key results?
I see three major takeaways:
- RNA-Seq is now about as inexpensive as microarrays, and comes with the advantages of higher reproducibility and a broader dynamic range. There are now few cases in which it makes sense to use microarrays over high-throughput sequencing technology (the main one that comes to mind is for comparison with legacy data, and even there I would argue that it is possible to effectively compare RNA-Seq and microarray data.)
- All of the sequencing platforms are producing high-quality, consistent data. Illumina, PacBio, 454, and Lifetech sequencing data showed a high (0.83) correlation coefficient for normalized transcript levels. That’s just slightly lower than what they saw when comparing each machine to itself (0.86). Considering that all of these platforms use entirely different underlying technologies, both for library preparation and the sequencing itself, this is powerful evidence that RNA-Seq is reliable and reproducible.
- Library prep methods matter more than you might think. Both poly-A-enriched libraries and ribosomally-depleted libraries show comparable detection of differentially expressed genes. But there are differences. Ribo-depleted libraries appear to have less coverage bias, yet they also showed 40-50% of the reads coming from introns, so that means a lot more sequencing to achieve the same depth of coverage.
We’ve seen the same thing at Cofactor in our our own internal research and development:
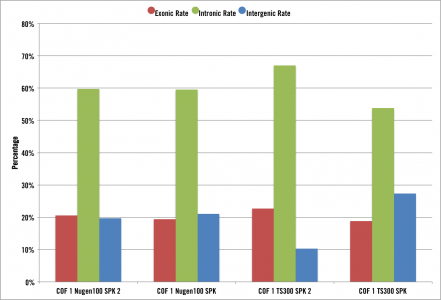
The above graph shows the same sample prepped with 2 different ribosomal depletion library kits and 2 technical replicates each. In both protocols, the majority of the reads mapped to introns.
Want to learn more?
There is a lot of great work here, and I’ve only touched on the highlights. If you’d like to take a more in-depth look, James Hadfield, one of the authors of the SEQC study, added some excellent additional commentary on both papers. RNA-Seq Blog wrote a brief introduction to the ABRF paper, and Nature Biotechnology offers their own take as well.
In our experience, we’ve seen the best results come from careful attention to every step of the process, from sample quality assessment to molecular spike-in controls to saturation curves — before analyzing the data for biological insights. It’s great to see the RNA-Seq community talking about these key issues, and these papers show how much progress has been made in just the last few years.
Links to the papers
- Detecting and correcting systematic variation in large-scale RNA sequencing data
- Multi-platform assessment of transcriptome profiling using RNA-seq in the ABRF next-generation sequencing study
- Normalization of RNA-seq data using factor analysis of control genes or samples
- The concordance between RNA-seq and microarray data depends on chemical treatment and transcript abundance
- A comprehensive assessment of RNA-seq accuracy, reproducibility and information content by the Sequencing Quality Control Consortium