Ribosomal RNA (rRNA) constitutes a large majority (> 97%) of the RNA species in any total RNA preparation. Therefore, to uncover the complexity of the < 3% mRNA and non coding RNA in a sample it is important to first deplete rRNA. Poly-A enrichment and probe based ribosomal RNA removal are by far the most popular methods currently in use. The scope of the experiment and the tissue type will primarily determine which rRNA depletion method is appropriate and is discussed in detail here. Now here’s something most RNA-sequencing providers don’t tell you at the beginning of your project. You can never achieve 100% rRNA removal. There are certainly measures one can take to avoid losing a significant portion of the sequencing data to rRNA reads but there is always some amount of rRNA carryover that is unavoidable. Therefore, when planning your RNA-seq experiment, always budget a few extra reads to account for those rRNA reads that come along for the ride. Depending on the depletion strategy this may range from 1-2% rRNA at the low end all the way up to 35% of the data or higher (this high of a percentage never happens at Cofactor but within the realm of possibility). At Cofactor, we report the ribosomal content front and center in your ActiveSite interface so you’re aware of the potential impact.
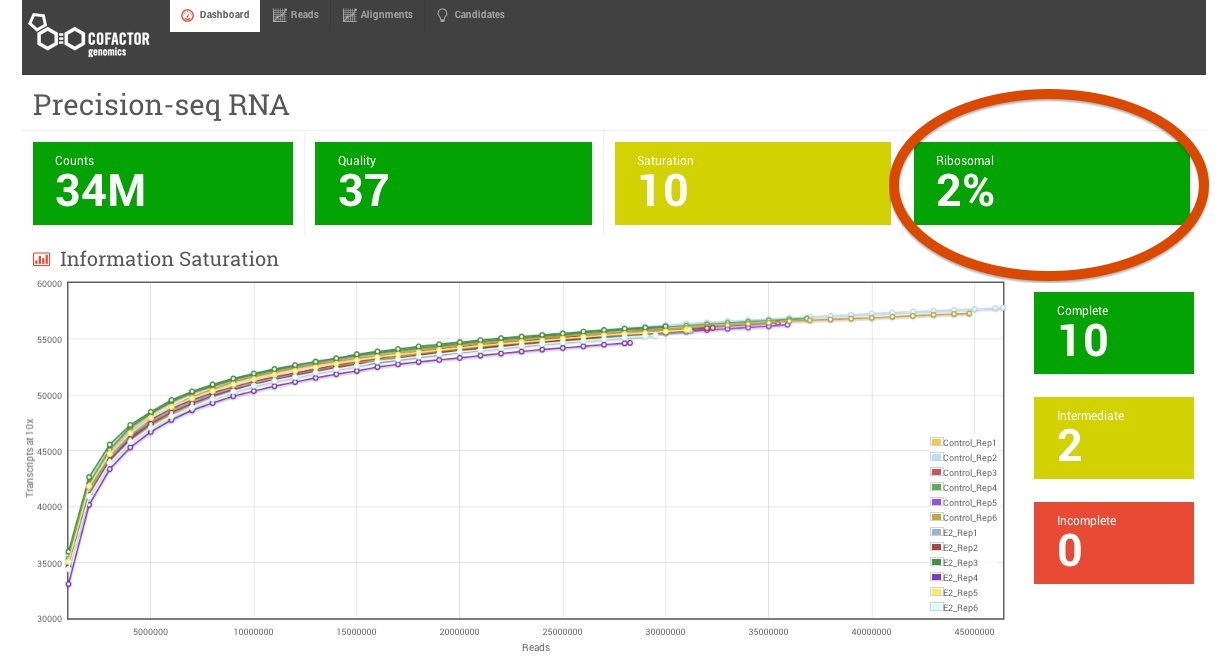
Ribosomal content is highlighted front and center in Cofactor’s ActiveSite
On the analysis side, ribosomal and mitochondrial content will normally have to be filtered out so that differences in rRNA reads across samples do not affect alignment rates and skew subsequent normalization of the data. This is relatively straightforward so rRNA depletion strategies are really aimed at getting a better bang for your buck for an RNA-seq experiment rather than improving the data itself. Why is all this rRNA hanging around despite all measures to get rid of it? The reasons are both technical and biological. Probe based methods suffer the technical challenge of requiring a pretty exact match of the organism’s ribosomal RNA and mitochondrial RNA sequences to the probe set. There just aren’t enough probe sets out there to match every single organism’s rRNA so we have to make do with something that’s close enough. Even for human or mouse samples where there are exactly matched probe sets, the hybridization will still fail to remove all rRNA. Depending on the source of the sample, it may be a complex mixture of a few different organisms (think gut lining with lots of bacteria for example) and unless all those rRNA species are targeted specifically for depletion, the rRNA will be present in the sequencing library. Poly-A enrichment is generally better at removing ribosomal RNA but a small percentage of ribosomal RNA can stick to the enrichment beads non-specifically. Sometimes Poly-A libraries contain rRNA due to the little known fact that ribosomal RNAs and mitochondrial RNAs are also polyadenylated.
We take all of this and more in to account when we design experiments for our customers. It’s all part of our aim to give you access to one of the most experienced teams in sequencing. If we can help you with your project, get in touch.
P.S. If you like this post, you might enjoy Cofactor’s newsletter. Receive each new post delivered right to your inbox! Sign up here.