How Low Can You Go?
Advantages of low-input RNA-seq over single-cell RNA-seq
Do you believe RNA sequencing has become an essential tool in addressing biological questions? That it is a powerful means for understanding phenotypic differences? We do! And that’s probably not surprising coming from an RNA-seq company. Traditionally, RNA sequencing refers to poly(A) RNA-seq or ribo-depleted RNA-seq (whole transcriptome). These approaches measure the average expression level for each gene across a large population of cells, often from a bulk biological sample in the form of a tissue section or pellet of cultured cells. These samples can be heterogenous, likely comprised of multiple cell types at different stages of development, apoptosis, necrosis, or spatial differentiation.
To address the heterogeneity of these materials, and truly understand the cell-to-cell variability in a sample, many researchers have turned to single-cell RNA sequencing. This technology is still fairly new, first published in 2009 (Tang, et al. Nat. Methods 6 (5): 377–82), and a few reagent and instrument manufacturers have built their own commercially-available kits and platforms including Fluidigm,10x Genomics, BioRad/Illumina, and BD.
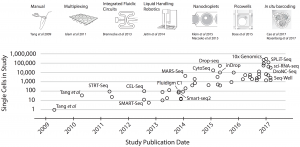
Image from: arXiv:1704.01379v2 [q-bio.GN]
Single-cell RNA-seq (or scRNA-seq), as the name implies, enables the measurement of the expression level of (a subset of) genes in an individual cell. Most often, a population of hundreds to thousands of cells is measured, and the distribution of expression levels from each gene are evaluated for the whole. While this technology has found applications in certain areas (identification of cell types, measuring heterogeneity/variability, etc), there remain many challenges associated with quantifying single-cell expression levels. Alternatively, low-input RNA sequencing, using subsets of cell populations or micro-dissected tissue, can be an ideal approach to obtaining high-quality, interpretable comparative expression information.

Sources of variability
– Amplification (technical noise) – the amount of RNA present in a single cell is limited (1-50 pg, dependent on cell type), and the resulting cDNA must be amplified to generate a sequencing library. This can introduce bias and noise in the data. With low-input RNA-seq, there may still be some amplification required (depending on the amount of starting material), however, it is much less than scRNA-seq. Cofactor has optimized this input-to-amplification ratio in our picoRNA workflow to ensure as little amplification as possible is utilized.
– Gene ‘dropouts’ – due to the low amount of starting material with scRNA-seq, it’s possible for some genes (especially moderate to low-level expressed genes) to be randomly “missed” in the library preparation method, even if they are present in the cell. This is less likely to happen with low-input RNA-seq experiments as the pool of molecules is larger and much more diverse.
– Biological noise – even genetically identical cells, under identical conditions, display high variability in their gene and protein expression levels. In single cell experiments, this biological noise is quite large, and must be averaged across many cells to truly understand the cell population. This is in contrast to low-input RNA-seq, where the biological variability between cells is represented as the ensemble expression level of the sample.
Statistical Significance
– Number of samples – due to the high biological and technical noise in a scRNA-seq experiment, hundreds to thousands of individual cells must be used, and the data analyzed and integrated to draw statistically-meaningful conclusions. While the commercially-available protocols make use of both cellular and sample multiplexing, the cost of these sequencing experiments still far outweighs a traditional RNA-seq experiment. Further, the sheer amount of data generated can be overwhelming, and depending on the provider/technology being used, you may find there is limited informatics support available. Cofactor’s low-input RNA-seq approach includes our comparative-expression analysis and ActiveSite interface, where you can easily sort and refine your data – including p-values and CVs for replicate groups with >3 samples.
– Cost of sequencing – while the cost of sequencing continues to decrease, the reality is that even at relatively shallow depths (10k reads/cell), when studying hundreds to thousands of cells the sequencing alone can become cost prohibitive quite quickly. Using low-input RNA-seq, once can craft an experimental design that enables sequencing deeply and with multiple biological replicates for a reasonable cost. And, with Cofactor’s expertise in this area, we can ensure high success rates and quick turnaround times, further adding value to your research.
Sample Logistics
– Shipping and dissociation – For scRNA-seq, cells must be dissociated into viable single-cell suspensions which are then loaded onto the platform. This presents challenges from an outsourcing and shipping perspective, as ideally, the cells are freshly dissociated prior to loading. However, with low-input RNA-seq, the sample logistics are a bit easier. If using sorted cells, they may be sorted directly into lysis buffer and shipped; cells may be suspended in a Trizol solution and shipped; extracted RNA may be provided (in low volumes); or FFPE core needle biopsies/sections/slides may be submitted.
– Sample types – scRNA-seq may only be used on samples which result in viable single-cell suspensions. This limits its utility to cell cultures and fresh or frozen tissue. In contrast, Cofactor’s two options for low-input RNA-seq can accommodate high-quality (RIN>7 for picoRNA) or low-quality (DV200 > 30% for RNAmplify or RNAccess) samples. This enables low-input RNA-seq to be broadly applicable, for multiple sample types, sources, and storage times.
– Spatial context – with standard scRNA-seq all spatial information is lost in the dissociation process. However, by utilizing laser capture microdissection (LCM) on snap frozen tissues to sequester specific cell populations, low-input RNA-seq may be used to elucidate spatial profiles.
So, if you happen to have access to a cell-isolation and barcoding platform nearby, and your scientific question truly relies on collecting information at the cellular level, AND you have the statistical prowess to make sense of all the data you collect – you’re on the right track with scRNA-RNA-seq!
But if your experiment does not check ALL those boxes, and you’re instead interested in:
- sorting cell populations via FACS/flow cytometry
- capturing small regions of tissue via LCM
- processing very small numbers of cultured cells (or cells with low RNA content)
- saving money and maximizing the insight from your budget
then low-input RNA-seq may be a great approach to gain powerful transcriptomic data with excellent technical reproducibility and reduced biological noise.
Are you interested in better understanding the technical details or logistics of these protocols? Reach out to schedule a time to speak with one of our Project Scientists today.
Other Helpful links:
https://arxiv.org/abs/1704.01379
https://hemberg-lab.github.io/scRNA.seq.course/introduction-to-single-cell-rna-seq.html
