The central purpose of an RNA sequencing (RNA-seq) experiment can be any of the following: to identify the structure of transcribed genes, explore splice junctions, characterize small RNA, identify novel transcripts and transcriptional start sites or to quantify transcription. For transcriptome-based studies, RNA-seq libraries are generated by the synthesis of double stranded cDNA followed by the addition of sequencing adapters. This method however, does not retain any information about the DNA strand from which the RNA was transcribed.
So what is the advantage of having strand specific sequence information? Apart from providing an insight into antisense transcripts and their potential role in regulation and strand information of non-coding RNAs, it aids in accurately quantifying overlapping transcripts. The last one is particularly relevant in organisms like prokaryotes and lower eukaryotes that have a more compact genome. With its applications growing, directional mRNA sequencing is gaining popularity. Computational analysis may help in retrieving strand information by exploring open reading frames, but this can be a tedious process. On the library end, several protocols and applications are now available that help keep strand specific information.
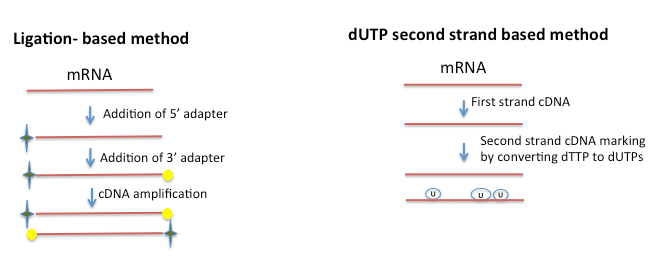
What we have learned through Cofactor’s R&D initiatives is that strand information can be retained either by specifically priming the 3′ and 5′ end of RNA (ligation based method), or by chemically modifying the 5′ and 3′ ends. Yet another approach marks the second strand by replacing the dTTPs with dUTPs and later separating the two strands using an adapter. This is commonly referred to as the “dUTP” based method. Since chemical modification is through a bisulphite modification, both library construction and analysis can be a little more challenging. The ligation based method stemmed off from the miRNA sequencing protocol of using the structure of RNA to specifically prime the ends and is better suited for transcripts <150bp while the dUTP technique is preferable for larger transcripts. Several protocols are now available for the ligation-based method and dUTP based method. They are fast, reliable and allow multiplexing. Ligation-based methods also allow for the elimination of ribo depletion steps that normally precede RNA-seq. Since priming of the 5′ and 3′ end and amplification result in enrichment of specific mRNAs it is possible to use total RNA as the starting material for library construction.
Will strand specific sequencing eventually replace conventional RNA-seq protocols? Maybe not. Conventional protocols are still the method of choice for most RNA-seq applications, being used in more than 90% of the RNA projects Cofactor has completed in recent quarters. Strand specificity data adds significant time to downstream computational analysis and this additional complexity may not be useful in projects not focused on overlapping transcripts. At Cofactor, strand specific RNA-seq is one of the solutions that has come out of our R&D initiatives to provide our customers with the right protocols for their projects. It is not recommended for every RNA project, but it is our scientists’ mandate to evaluate new approaches as they come online. By understanding project goals, our scientists can make sure that you’re using the best protocols while making sure you’re not wasting time or money. At Cofactor it’s our job to keep up with the new trends so you can get on with discovery.
