
Cofactor recently presented a poster at AGBT 2016, in collaboration with NuGEN, and I thought it might be interesting to share it here (in a blog format) for those of you who were not able to attend the conference. You will see below that this is a methods poster, in other words, we wanted to test and confirm a method to interrogate these molecules from exosome samples. We believe we have accomplished that and for next steps we look forward to better understanding what transcript and gene differences exist between the renal cancer and normal exosome samples. Here is a link to the product we used: NuGEN Ovation Single Cell RNA Seq System. Enjoy!
A Method for Surveying the Long-RNA Landscape of Exosomes
Jon R. Armstrong1, Jeff Hiken1, Ajay Khanna1, Stephanie Huelga2, Luke Sherlin2 and Doug Amorese2
1. Cofactor Genomics, St. Louis, MO
2. NuGEN, San Carlos, CA
Introduction
The emergence of next-generation sequencing platforms has enabled the discovery and interrogation of RNA from tissues, biofluids, and extracellular vesicles at remarkable levels. Recently, interest has grown in sequencing RNA found in extracellular vesicles called exosomes, because of their role in cell-cell communication and immunoregulatory processes [1]. The contents of exosomes have been shown to consist of multiple RNA types including, micro RNA (miRNA) and messenger RNA (mRNA), as well as other biologically active molecules [2, 3]. Extensive work has focused on assessing the miRNA content of exosomes and it’s role as a putative disease biomarker. However, other types of RNA, such as long-RNA (> 200 bp; long non-coding RNA [lncRNA], mRNA, and circular RNA [circRNA]), may play an equal or more important role in cellular communication and cause alterations to biological pathways that affect disease development. Long-RNAs are a minor proportion of the RNA material in exosomes and until recently, it was extremely challenging to generate sequencing libraries from the very small amounts of long-RNA extracted from exosomes. However, new methods have emerged, such as NuGEN’s Ovation® Single Cell RNA-Seq System, which enable a single library to be generated from extremely small amounts of long-RNA. Unlike poly(A) library methods, these libraries retain the full complement of long-RNA species (e.g. lncRNA, mRNA, and circRNA). In order to test the Ovation Single Cell RNA-seq System as an applicable method for generating next-generation sequencing libraries, consisting of multiple long-RNA species, and explore the population of long-RNA molecules in exosome samples, we applied the Ovation Single Cell RNA-Seq System to total RNA extracted from exosomes. The libraries were sequenced and the resulting data was analyzed to characterize the landscape of exosome derived long-RNA species. Here, we present the method workflow, number and types of observed long-RNA’s, relative proportions of long-RNA’s across multiple control and renal cancer exosome samples and propose functional categories for genes enriched in exosome samples.
Materials and Methods
Sample Procurement
Normal control (n=3) and renal cancer (n=3) serum samples from separate individuals were purchased from Bioserve (Beltsville, MD). Normal control and renal cancer serum samples were matched, based on meta-data, as closely as possible prior to ordering. Samples were stored at -80 ˚C before exosome isolation.
Exosome and RNA Isolation
1 – 3 ml of serum was passed through 0.8 µm syringe filters (Millipore Millex-AA [cat. no. SLAA033SB]) to remove cell-membrane debris. Exosomes were then isolated using the exoRNeasy Serum/Plasma Maxi Kit (Qiagen, Valencia, CA), according to manufacturers directions. Briefly, exosomes were bound to a column matrix and washed. Exosome RNA was eluted from the matrix with Qiazol, and then isolated using RNeasy MinElute spin columns. RNA concentrations were quantitated using an RNA 6000 Pico assay run on the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA).
Library Generation
NGS libraries, for paired-end sequencing on the Illumina sequencing platform, were prepared using the Ovation Single Cell RNA-Seq System (NuGEN Technologies, San Carlos, CA). Illumina-compatible barcoded adaptors were included in the kit. After adaptor ligation, libraries are pre-amplified with 14 PCR cycles (Library Amplification I). For 10 ng exosome RNA input, libraries were further amplified with 10 PCR cycles (Library Amplification II).
Sequencing and Analysis
Libraries from each sample were multiplexed and sequenced on the NextSeq 500 instrument (Illumina, San Diego, CA) as paired-end 150 base reads (2×150). The resulting reads in FASTQ format were aligned to GENCODE v24 lncRNA and protein-coding RNA transcript sets. Alignments were performed using Novoalign (version 3.02.05; Novocraft, Malaysia) in “-r all” mode to allow reads to align to multiple transcripts. RPKM values for the alignments were calculated from the BAM output files, using the standard RPKM formula, with a custom Perl script. Reads were also aligned to the hg19 reference genome using STAR (version2.4.1c) with option “–outFilterMultimapNmax 1” to only allow unique alignments [4]. Transcript RPKM values for each sample were loaded into Cofactor Genomics’ ActiveSite Differential Expression Viewer to derive aggregate and unique transcript counts. The software program Known and Novel Isoform Explorer (KNIFE) was used for circRNA detection and run according to the author’s recommendations [5]. The following command line was used: completeRun.sh $PWD/reads complete $PWD thisproject 13 sam circReads 67 1 none. RNA-SeQC was used to generate read designations for each sample [6]. Database for Annotation, Visualization, and Integrated Discovery (DAVID) (version 6.7) was used to identify gene functional annotation terms that were significantly enriched in genes shared across all renal cancer samples [7]. A list of gene symbols was generated for the aggregate dataset and was used as input into DAVID.
Results
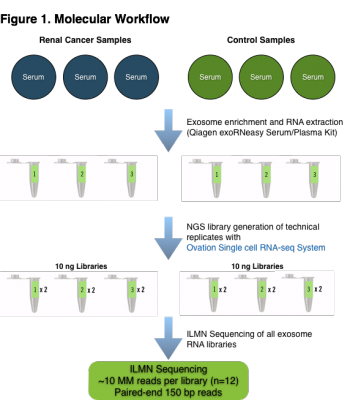
Molecular Workflow. Serum samples from renal cancer and normal control individuals (n=6) were enriched for exosomes and total RNA was extracted in order to investigate the efficacy of the NuGEN Ovation Single Cell RNA-Seq System for interrogating the composition of long-RNA molecules from exosomes. The NuGEN Ovation Single Cell RNA-Seq System is a random priming library method which will prime and produce cDNA from many types of RNA species in a sample. Duplicate technical replicate libraries were produced from each sample to investigate the variability induced by the Ovation Single Cell RNA-Seq library protocol. 12 total libraries were generated from 6 samples and the libraries were sequenced to ~10 million reads each on the Illumina NextSeq 500 platform using paired-end 150 base reads.
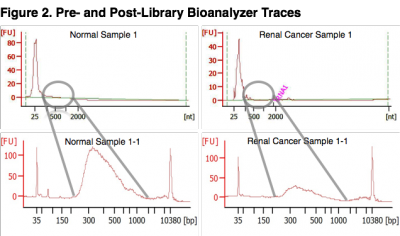
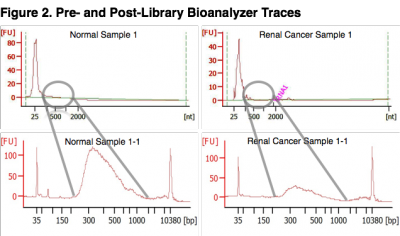
Pre- and Post-Library Bioanalyzer Traces. The top two panels represent bioanalyzer traces from two samples following RNA extraction. The bottom two panels represent bioanalyzer traces after final library amplification. Exosomes were isolated from each sample, the RNA extracted and yield measured. Serum samples yielded an average of 22.6 ng/mL (SD = 8.1 ng) of total RNA from exosomes. Sequencing libraries were generated using 10 ng of exosome total RNA input to the NuGEN Ovation Single Cell RNA-Seq System. The bioanalyzer traces for replicate libraries exhibited equivalent topologies (not shown) demonstrating that the Ovation Single Cell RNA-Seq library method was robust at a 10 ng total RNA input amount. The input amount of long-RNA (> 200 bases) was likely underestimated since a large proportion of the measured concentration consisted of small-RNA species, identified by the large spike (< 100 bases) at the beginning of the bioanalyzer traces. The NuGEN Single Cell System employs a magnetic bead cleanup step which removes adapter ligated species smaller than 200 bases. Thus, cDNA inserts larger than approximately 70 bases were retained for sequencing (Illumina adapters add 130 bases to the inserts). miRNA species were removed by the bead cleanup.
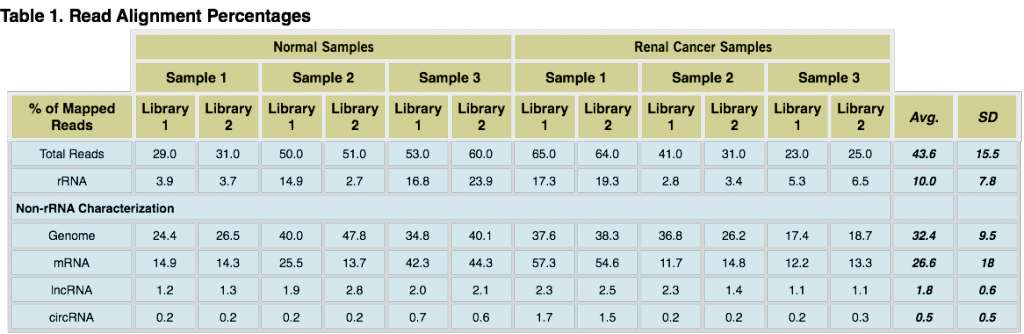
Read alignment percentages. The table above presents the percentage of read alignments for each library and RNA species (Genome = hg19 alignments; mRNA = GENCODE protein coding database alignments; circRNA = detection using KNIFE software; lncRNA = GENCODE lncRNA database alignments) including averages (Avg.) and standard deviations (SD). Although the percentage of read alignments across samples was not highly correlated, the percentage alignment between replicates was largely comparable. Differences in the RNA content of exosomes is to be expected, however technical replicate libraries should exhibit nearly the same sequencing metrics (as seen here) if the molecular steps in a chosen library method are robust.
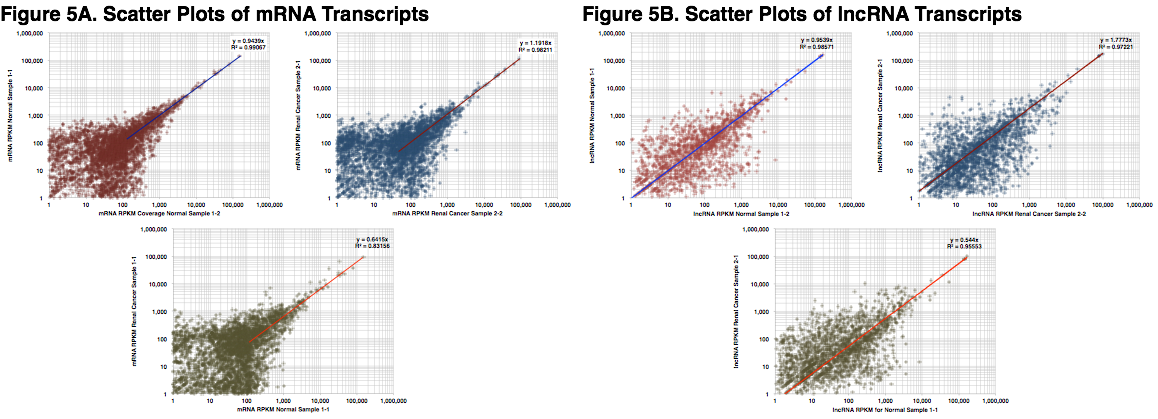
mRNA and lncRNA Transcript Scatter Plots. Scatter plots were generated, using RPKM, for mRNA and lncRNA species to investigate the correlation and molecular noise between replicate library samples. The slope and coefficient of determination (R2) were calculated for each comparison. The plots in Figure 5A were generated using RPKM signals of protein-coding alignments from Normal (red) and Renal Cancer (blue) samples. The lower plot (green) displays the slope and R2 between Normal Sample 1-1 and Renal Cancer Sample 1-1. Replicate libraries exhibit excellent linear reliability while, as expected, the plot of Normal Sample 1-1 and Renal Cancer Sample 1-1 exhibits less linear reliability (a greater number of differentially expressed coding transcripts). Figure 5B shows the same plots for lncRNA alignments. The above plots exhibit higher levels of noise than might be seen when comparing RNA sequencing data from tissue samples. However, exosomes originate from many different tissues and also exhibit a temporal component. Thus, higher levels of noise may be inherent with these types of samples. In addition, sampling noise could arise during the sequencing process (i.e. low number of reads per sample) or molecular noise from the Ovation Single Cell System at the current input amount (10 ng). These sources of noise could be attenuated by generating additional sequencing reads for each sample and/or increasing the amount of long-RNA input to the molecular method.
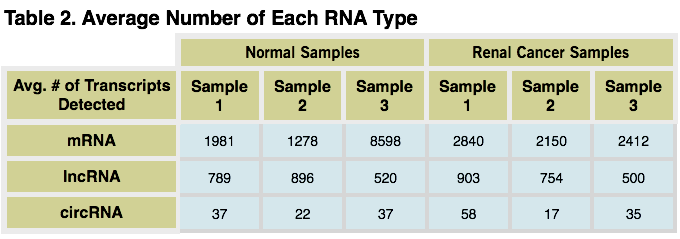
Average Number of Each RNA Type.The average numbers of each RNA type, from normal and renal cancer samples, are shown in the table above. The average was calculated from the summed number of signals in replicate libraries. A lower RPKM threshold cutoff was employed to count only high confidence signals (mRNA = 100, lncRNA = 500, circRNA = 5).
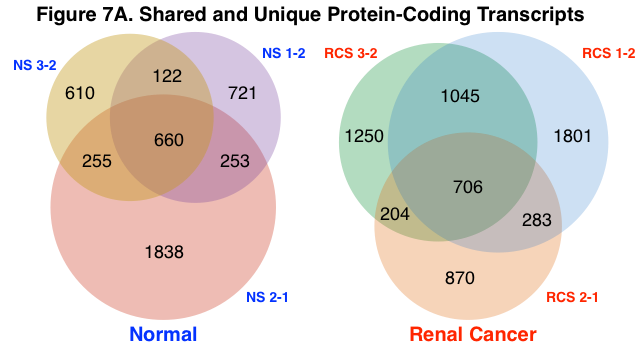
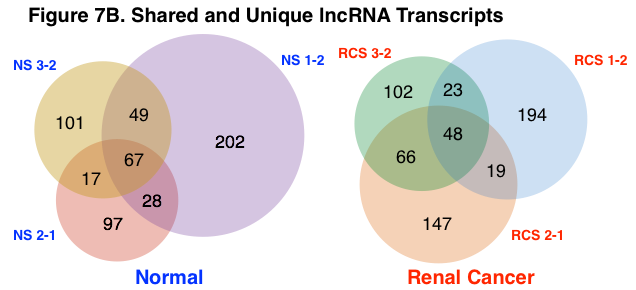
Shared and Unique Protein-Coding and lncRNA Transcripts. The number of aggregate and unique protein-coding and lncRNA transcripts were calculated across normal and renal cancer sample groups using a lower RPKM threshold cutoff of 100 and 500, respectively. Figure 7A represents the number of aggregate and unique mRNA transcripts for normal and renal cancer samples. Figure 7B represents the number of aggregate and unique lncRNA transcripts. Normal samples shared a greater percentage of both protein-coding and lncRNA transcripts than renal cancer samples (normal – protein-coding = 33%, lncRNA = 27%; renal cancer – protein-coding = 30%, lncRNA = 18%). The detection of circRNA species was determined using a separate analysis algorithm (KNIFE), thus the number of shared and unique circRNA species was not calculated.
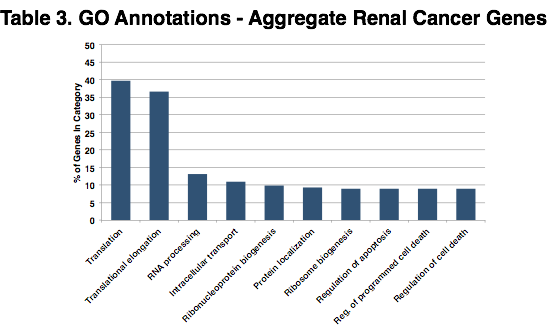
Gene Ontologies for Aggregate Renal Cancer Genes. The chart above shows the top 10 significantly shared gene ontology (GO) terms across the aggregate group of renal cancer genes. The 706 transcripts (including isoforms) shown above in Figure 7A – Renal Cancer, represent 191 genes which were used as input to DAVID. Over 75% of the coding transcripts in these exosome samples were involved in translation and elongation functions.
Conclusions
- Exosomes contain an abundance of long-RNA species including protein-coding genes (mRNA), lncRNA, and circRNA.
- The Ovation Single Cell RNA-Seq System is an effective and robust method for generating next-generation sequencing libraries consisting of long-RNA from exosomes.
- mRNA, lncRNA, and circRNA can be identified and analyzed from a single library preparation using multiple data analysis pipelines.
- Numerous genes are shared across exosome samples, with the largest proportion making up translation and translational elongation functions.
References
- Théry C., Ostrowski M., Segura E. Membrane vesicles as conveyors of immune responses. Nature Reviews Immunology. 2009;9(8):581–593.
- Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007:9(6):654–9.
- Schorey JS, Bhatnagar S. Exosome Function: From Tumor Immunology to Pathogen Biology. Traffic (Copenhagen, Denmark). 2008;9(6):871-881.
- Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15-21.
- Szabo L, Morey R, Palpant NJ, Wang PL, Afari N, Jiang C, Parast MM, Murry CE, Laurent LC, Salzman J. Statistically based splicing detection reveals neural enrichment and tissue-specific induction of circular RNA during human fetal development. Genome Biology. 2015, 16:126.
- Deluca DS, Levin JZ, Sivachenko A, Fennell T, Nazaire MD, Williams C, Reich M, Winckler W, Getz G. (2012) RNA-SeQC: RNA-seq metrics for quality control and process optimization. Bioinformatics (2012) 28 (11): 1530-1532.
- Glynn Dennis Jr., Brad T. Sherman, Douglas A. Hosack, Jun Yang, Michael W. Baseler, H. Clifford Lane, Richard A. Lempicki. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biology 2003 4(5): P3


{kind=link}