Picking up where we left off in our last post, the first problem we see with current RNA-seq is the unstructured question of “How many reads do I need?” There’s constant discussion on what the right number is for a reliable RNA-seq project. Many researchers choose a number based on a recent publication or work from a colleague down the hall. Few are determined on a quantitative measure.
But there’s no reason to guess. We as biologists readily acknowledge that each sample, time point, and phenotype have different RNA characteristics. Also depending on what specific process or pathways we are interested in we may need to sequence further into the RNA pool (or less) in order to adequately characterize/quantify that portion of RNA that we are most interested in. It is possible to gain much further confidence in the level of characterization of the RNA component of the sample by calculating a discovery/saturation rate for a given experiment.
At Cofactor, by taking the expression profile of a given RNA-seq experiment (fig1) we are able to calculate a signal/noise cutoff. Once that signal/noise cutoff is determined we are able to use a saturation analysis (fig2) to calculate a discovery rate. This curve as well as the discovery rate becomes both a qualitative and quantitative measure of how far we have sequenced into the RNA pool. Based on past deep sequencing RNA-seq experiments we are able to make an assessment to what extent the transcriptome has been characterized for a given sample (using a scale of 1 to 10, where 10 represents full saturation).
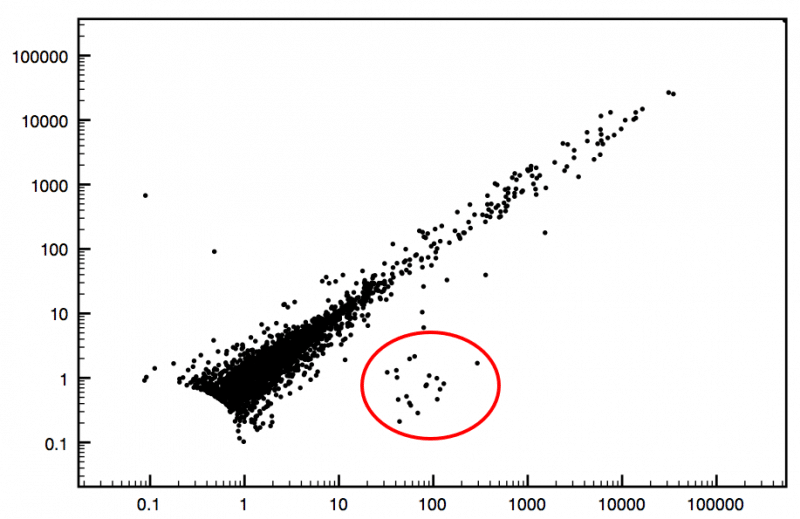
Figure 1

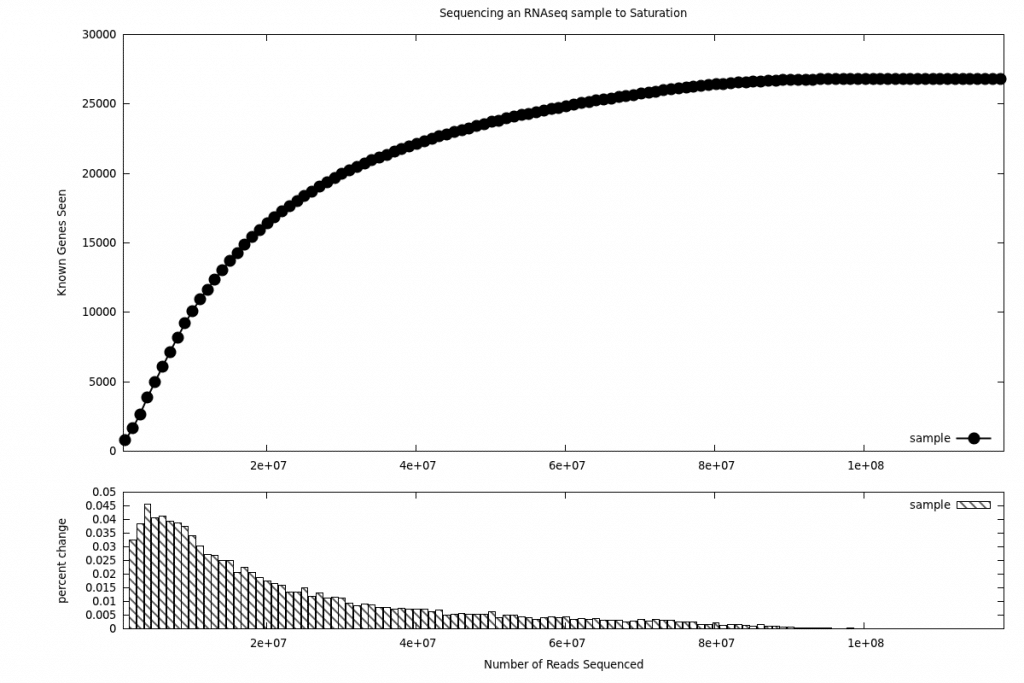
Figure 2
This feedback between discovery rate and read count gives important perspective and/or confirmation to what level the RNA component has been characterized. Leaving researchers confident at the start of their experiment that they are getting the appropriate number of reads.
Here’s the next installment of our RNA-seq DX series.
